Beyond MiModD: Things the package cannot do for you¶
Sequenced reads quality control¶
MiModD does not include any tools to check the quality of your sequenced reads input files. If quality control is not done for you by the sequencing facility providing the reads, we recommend the free and open-source FastQC fastq data quality control tool, which requires only Java on your system and offers a simple user interface, extensive functionality and good documentation.
If the quality control reveals serious problems with your input you may consider using a tool like FastX to filter or trim reads based on quality and/or consult your sequence provider for other options like resequencing your samples
Indel realignment¶
MiModD does not (currently) include an indel realignment step before variant calling. Local realignment of regions around indels is a widely accepted method to increase the accuracy of calling indels at the variant calling step.
Since indels, typically, represent only a small fraction of the total variants in mutagenized model organism genomes (the vast majority of variants are single nucleotide changes), including such a step in MiModD is not one of our priorities.
If you want to include indel realignment in your analysis workflow, we recommend using the RealignerTargetCreator and IndelRealigner tools from the GATK toolkit on the aligned reads file produced by the snap tool.
See also
- the GATK RealignerTargetCreator tool documentation
- the GATK IndelRealigner tool documentation
Alignment visualization¶
MiModD does not offer any alignment browser tool.
Once you have obtained a list of bona fide variants, it is always a good idea to go back to the aligned reads file and check manually how well the alignments in the affected region support the variant. A graphical view of the aligned reads stacked on top of the reference genome can often be used to rule out false-positive variant calls quickly. This applies, in particular, to indels and deletions, for which the rate of false-positive calls produced by MiModD is relatively high (for deletions, in fact, the vast majority of calls will be false-positive).
Our preferred alignment browser is the Integrative genomics viewer IGV available for download from the Broad Institute.
IGV is free software requiring a one-time registration to download. It requires Java to run and an internet connection to download annotated reference genomes.
To visualize MiModD-generated alignments in IGV, all you need is:
- the aligned reads file generated by the snap tool and
- a corresponding bam index file, which you can generate with the index tool or will be available for download along with the bam file from the Galaxy interface
Annotated reference genome files for almost any model organism can be downloaded directly through IGV.
The following two figures illustrate the power of visual variant inspection using as examples two regions on C. elegans chromosome III, for which the delcall tool reported deletions in an analysis.
Figure 1 shows the IGV stacked alignment view of the first region, a predicted ~100 bp deletion with a delcall score of 87:
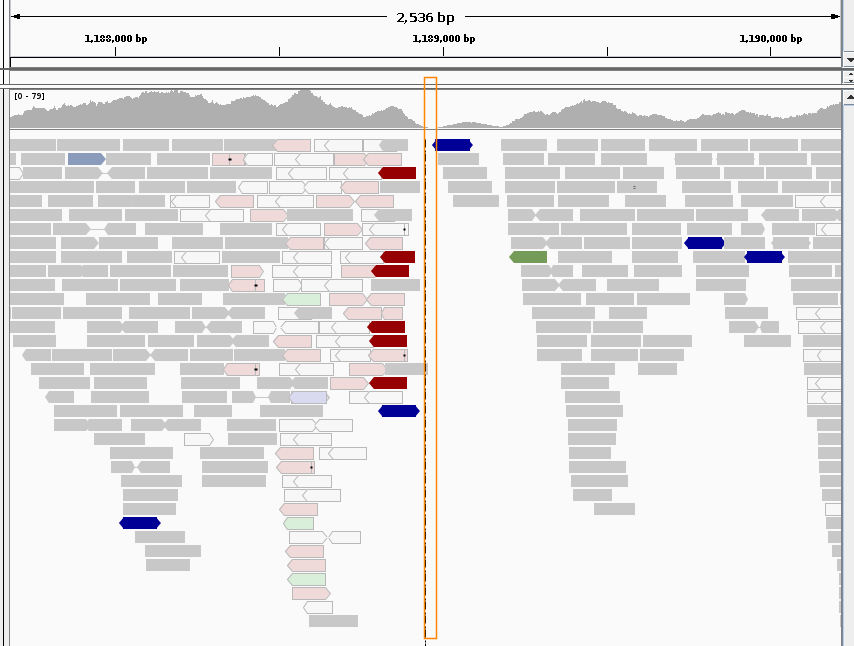
From this it is quite apparent that the predicted deletion (orange rectangle) lies in a poorly aligned region indicated by low-quality mappings (white and light-colored reads) and by various kinds of pair-orientation problems (colored reads). While this does not rule out some kind of complex structural change to the chromosome in this region, this will normally not be a high-priority candidate variant to follow up experimentally.
In contrast, figure 2 shows an IGV view of the second region with a predicted ~250 bp deletion with a delcall score of 52:
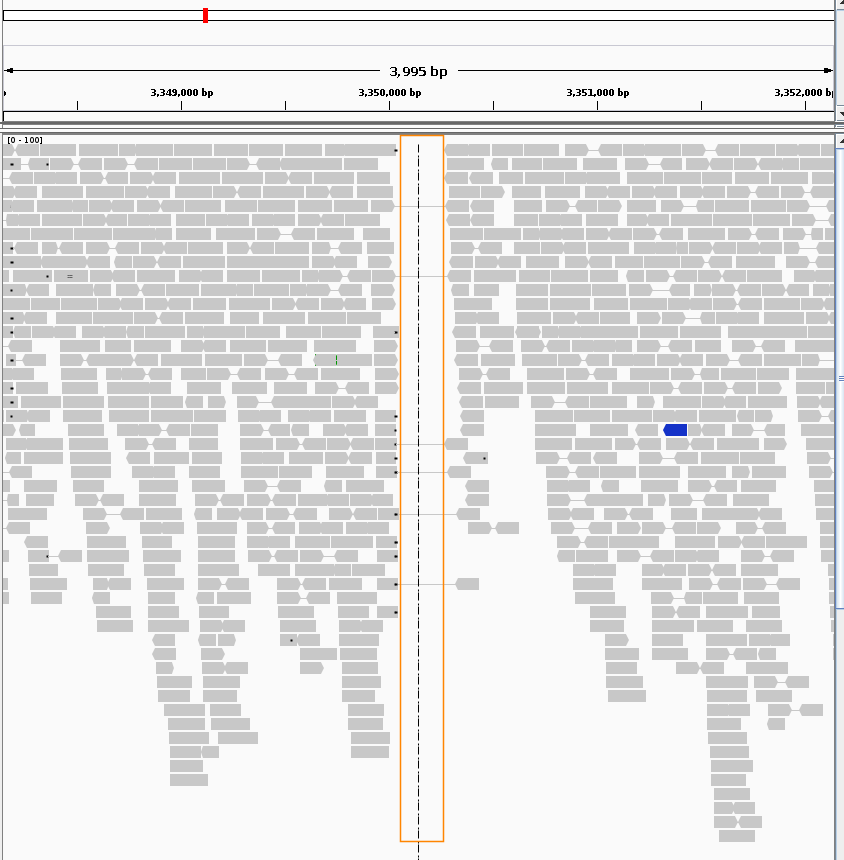
Even though the delcall-reported score for this second candidate is lower than for the first one, the visual inspection reveals it to be a far better candidate. The uncovered region is sharply delineated and resides in the center of an otherwise perfectly aligned region. In addition, the read pairs spanning the uncovered region show no sign of mismapping, but instead have good mapping quality and normal read orientations.