MiModD 0.1.9 Documentation¶
What is MiModD ?¶
MiModD is a comprehensive software package for the mapping, identification and annotation of phenotypic mutations in model organisms from whole-genome sequencing (WGS) data.
Whom is it for ?¶
MiModD was designed specifically around the analysis needs of biologists/geneticists with limited bioinformatical knowledge, is really straightforward to install and easy to use (in particular, when integrated into a local Galaxy instance).
Analyses can be performed on anything from single-user off-the-shelf desktop computers and notebooks (provided they run Linux or macOS and meet some minimal hardware requirements) to department- or institute-level WGS data analsyis servers. When combined with Galaxy it can even turn your regular PC into a small analysis server for your research group.
Alternatively, MiModD is now offered on some public Galaxy servers so, if you do not mind high-volume sequence data transfers, you can start using it immediately.
MiModD could be interesting for you if:
you are a geneticist, who wants to identify mutations in a mutant strain of a model organism.
You probably have a good understanding of the principles of mutation mapping, and you know that whole-genome sequencing can speed up this process enormously, but you do not know how you would analyze WGS data and you do not know a bioinformatician who could do it for you.
Alternatively, you may have a bioinformatics facility in your institute, but they are overwhelmed with user requests and have a long queue and limited time per experiment so you would prefer to analyze your data independently.
you are analyzing WGS data already yourself, but you think your own computer cannot do such computation-intensive tasks. So you are using web-based tools, but it is really annoying to transfer all those huge files back and forth over the internet.
you are working in a bioinformatics facility and you are overwhelmed with user requests and would like to leave relatively standard tasks to your clients to have more time to tackle challenging problems.
MiModD is beginner-friendly, but that does not mean that it produces simplified results. Internally, it uses established tools like the spaced-seeds aligner SNAP and samtools/bcftools for variant calling, which ensures quality output.
Package Highlights¶
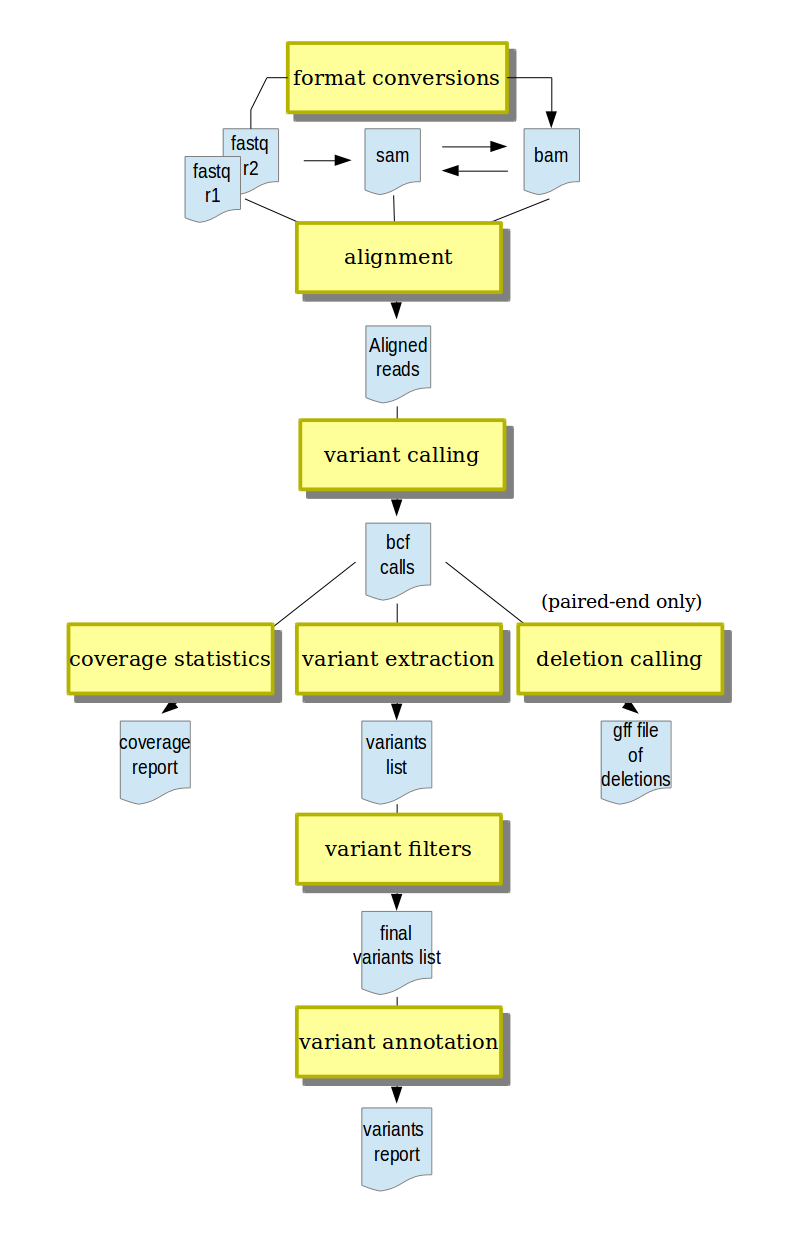
A typical analysis workflow in MiModD.
Functionality¶
- short-reads alignment from different input formats (fastq and gzipped fastq, SAM, BAM)
- conversion between input formats and annotation with run metadata
- variant calling (SNPs and indels)
- variant post-processing, including filtering and annotation
- deletion calling (currently only for paired-end data)
- linkage analysis for variant identification through mapping-by-sequencing approaches
User-friendliness¶
- completely automated management of temporary files
- analysis of multiple samples through a single linear workflow
- built-in multithreading in the alignment and variant calling steps for increased speed
- choice between a consistent command-line interface and a complete and straightforward integration of the package into a Galaxy server
- linkage analysis support for several different mutation mapping schemes
- publication-quality plots of linkage data (similar to the output produced by the earlier and now unmaintained CloudMap suite of tools that MiModD can replace)
Outline of this User Guide¶
See also
The complete Table of Contents
-
This section describes the installation of MiModD and all required and optional software including a guide to installing Galaxy and integrating MiModD into it.
-
We hope you will find MiModD and, especially, its Galaxy interface beginner-friendly and easy to use. The package is, however, versatile enough to be compatible with a number of different use-cases, which is best explained to new users by taking them through some example analyses that introduce the functionality of MiModD step-by-step.
Mapping-by-Sequencing with MiModD
This section explains in detail how to use MiModD and its NacreousMap engine to analyze and visualize variant distributions and inheritance patterns in order to pinpoint mutations of interest.
-
This section holds the detailed description of the MiModD command line and Galaxy user interface. It presents and explains all tools of the package and the complete set of parameters that can be used with each of them.
-
We have started to compile a list of recipes for performing some typical tasks that you are likely to encounter as a user of MiModD. Not all of these are MiModD-specific, but there are also some Galaxy recipes for working efficiently with WGS datasets.
-
This section explains some of the peculiarities and possibilities (like Galaxy Workflows) that will concern you only if you are using MiModD through its Galaxy interface.
Beyond MiModD: Things the package cannot do for you
MiModD is almost a one-stop solution for analyzing WGS data. There are certain tasks, however, that we consider to be outside the scope of the package. This section focuses on things that you cannot do with MiModD and recommends third-party software for these tasks.
File Formats and Compatibility
An overview of the various file format specifications that MiModD recognizes and uses, and of the MiModD tools that can be used to investigate and manipulate them.
-
MiModD is aimed at end-users. It is NOT another Python package for manipulating next-generation sequencing data programmatically. That said, as a Python programmer you may still find certain parts of the package useful for your work, or you may wish to contribute code or bug-fixes. In these cases, this section may serve as your starting point for exploring the package code.