MiModD and Galaxy¶
Native support of Galaxy¶
Unlike most other bioinformatics software MiModD was written with Galaxy integration in mind from the start. What this means is that:
- you do not rely on third-party wrappers for Galaxy, but they are an integral part of your MiModD download. Every release of MiModD comes with updated Galaxy wrappers that reflect all changes in the command line tools, i.e., you always experience the latest functionality also from Galaxy.
- the MiModD tools are designed to be compatible with simple and efficient Galaxy workflows. The Galaxy wrappers do not have to go to great lengths to fit the software into the Galaxy framework - things just work.
- even if you do not know how to add tool wrappers to Galaxy, MiModD knows and can do it for you (see here for details on how to integrate a standard installation of MiModD into Galaxy).
- if you prefer, you can, of course, install MiModD directly from the Galaxy Tool Shed instead.
- MiModD respects Galaxy slots settings. Although MiModD provides its own configuration option for multithreading, if you configure Galaxy to assign a specific number of CPU slots to any MiModD tool, the tool will respect that setting leaving Galaxy administrators in full control over system load.
Usage suggestions for MiModD from Galaxy¶
Even though we try our best to ensure a good user experience when working with MiModD through Galaxy, there are a couple of peculiarities inherent to Galaxy that may take some getting used to for beginners. Though these issues aren’t really about MiModD itself, but rather about getting things done with Galaxy in general, we try to cover the most important ones in the following.
Setting up an Admin account on your Galaxy¶
Note
This will only be of importance to you, if you have are managing your own Galaxy instance, but not if you are working on any Galaxy server provided by others.
Many of our other suggestions below will require you to work from an Admin
account at least at one step. The procedure to set up such an account is
rather simple and described here in the Galaxy
documentation. One known caveat for beginners is that you need to
uncomment (i.e., remove the leading # if it is there) the
admin_users = ... line, or it will be ignored by Galaxy.
Getting large files into Galaxy¶
Generally, the Upload Data tool of Galaxy makes it easy to get data into your current history - until you try to upload a large file (> 2 GB), that is. If you try that you will get an error message that suggests to use a FTP client instead.
This is, in fact, the way to upload such data to a remote Galaxy server that supports this method, but if you are using MiModD on your own Galaxy server that you are running on your local machine, or if you are working with a department-level Galaxy, for which it is easy to provide the data on a local disk, then a Galaxy Data Library solution may be a better choice for you.
Hint
By uploading your data to such a library once you have it available for import into as many histories as you like without having to create additional copies of the data everytime.
Galaxy data libraries are a feature that is documented very well. In particular, there is
a section in that documentation about setting up Galaxy to use Data Libraries
A frequent mistake by beginners when following these instructions is that they forget to uncomment the
user_library_import_dir = ...line after editing it, i.e. they leave it with a leading#, which causes it to be ignored by Galaxy.Also, note that the upload directory configured on this line can be any directory accessible to Galaxy and does not have to be an ftp upload directory like in the example given in the documentation (so if you do not have one, never mind).
a section about working with Data Libraries.
Copying vs linking files
A very useful option to emphasize here is the Link files instead of copying option available in the Data Library Upload interface.
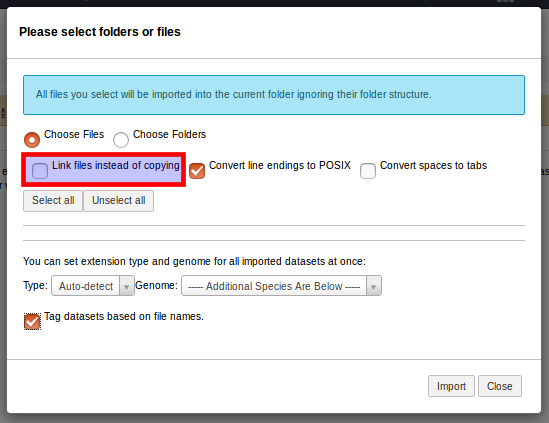
When you select this option, Galaxy will not make any internal copy of the dataset so selecting this is especially attractive for really huge file. Whenever you are using the dataset from any history later, what you will really be accessing is the original file in your upload directory.
Of course, the downside to this is that it is easy to break things. If you ever remove the file from the upload directory (forgetting that this is a linked file), all corresponding datasets in all histories will become useless!
Understanding Galaxy datatypes¶
In Galaxy, every dataset has metadata associated with it, which is just a word for things the Galaxy server knows (or, sometimes, thinks it knows) about the dataset. The most important piece of this information is the datatype of the dataset.
The most important thing you need to know about datatypes is this: every Galaxy tool, including the MiModD tools, informs the Galaxy server, which types of datasets it can deal with as input, and Galaxy will then make sure that only datasets that have an appropriate datatype will be offered as input choices in the user interface of the tool.
This means that, to work, e.g., with a MiModD tool that requires a reference
genome in fasta format, it is not enough that you have such a dataset
available in your history, but that dataset also needs to have its datatype set
to fasta or you will not be able to select it in the tool interface.
Normally, datatypes just work without you knowing about them, but to avoid problems it is still good practice to:
declare the datatype of a new dataset on upload;
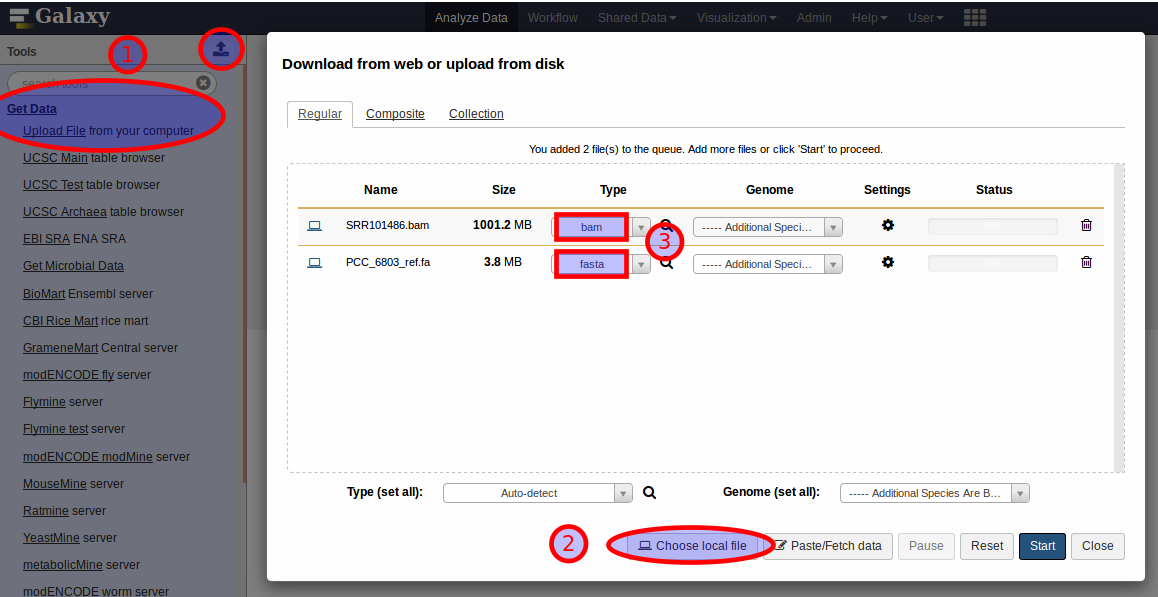
The upload interface of Galaxy provides selection lists (3) to choose the datatype of each new dataset from.
Leave it up to Galaxy to autodetect the type only if you are really unsure about what to select.
pay attention to the datatype associated with the datasets in your history
The datatype is shown in the expanded dataset view under format.
If a dataset that you would like to use with a tool is not offered as an input dataset, this format field should be the first thing to investigate.
make sure all your fastq datasets have a datatype of
fastqsangerunless you really know they are not in this format (compare https://galaxyproject.org/support/fastqsanger/); if the content is gzip-compressed the datatype should befastqsanger.gz.
In addition, it is helpful to understand that some datatypes have an
hierarchical relationship. For example, every tabular dataset also has an
implied datatype txt and every vcf dataset also qualifies as both
tabular and txt, but the reverse relations are not true. Thus, when a
particular tool asks for tabular input, you will be able to choose from any
vcf and tabular datasets in your history, but not from datasets of
the more general datatype txt, even if some of them are actually
tab-separated. This is the reason why you should try to select the most
specific datatype available for any new dataset on upload.
Viewing html datasets in Galaxy¶
As a security measure, Galaxy strips html datasets of all javascript and style information before displaying them to the user. This is done to guard against malicious html output generated by tools, which could serve as an attack vector, but it has the inconvenient side-effect that even the html output produced by harmless tools (like the MiModD Report Variants tool) does not look as intended by their authors anymore.
As a solution for this dilemma, Galaxy allows Admin users to manage a whitelist of installed tools for which no html filtering should be performed. This list is accessible from the Galaxy Admin interface as shown below.
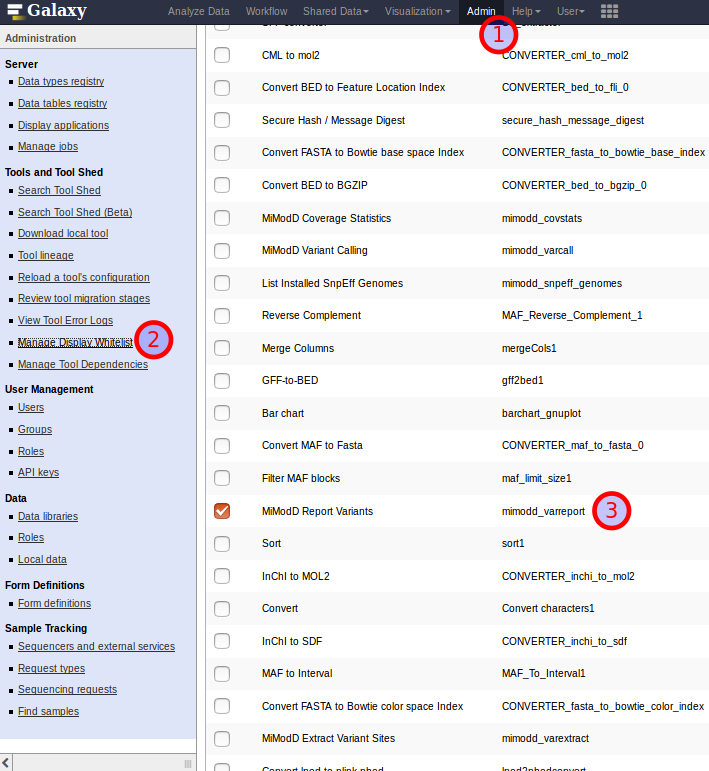
Three steps towards excepting the MiModD Report Variants tool from Galaxy’s html filtering.
Using MiModD on public Galaxy servers¶
For anyone interested in using MiModD in the cloud, we are maintaining a list of public Galaxy servers offering MiModD tools.
This page also has site-specific usage instructions that detail analysis steps that work differently on particular public servers than with a locally installed MiModD.
Description of MiModD workflows for Galaxy¶
Work in progress
We apologize, but this section of the User Guide still needs to be written.
For the time being, you may look at our general presentation of MiModD analysis workflows for mapping-by-sequencing type of experiments. While the discussion there is focusing more on the command line than on Galaxy, the information should not be that hard to transfer.